
More Information
Submitted: December 11, 2023 | Approved: December 28, 2023 | Published: December 29, 2023
How to cite this article: Danø C, Mikkelsen KV. Environmental Effects on the Norbornadiene-quadricyclane Photoswitch for Molecular Solar Thermal Energy Storage. Int J Phys Res Appl. 2023; 6: 203-215.
DOI: 10.29328/journal.ijpra.1001074
Copyright License: © 2023 Danø C, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Keywords: Electrocyclic reactions; Molecular photoswitch; Molecular thermoswitch; Energy storage

Environmental Effects on the Norbornadiene-quadricyclane Photoswitch for Molecular Solar Thermal Energy Storage
Christian Danø and Kurt V Mikkelsen*
Department of Chemistry, University of Copenhagen, Universitetsparken 5, 2100, Copenhagen Ø, Denmark
*Address for Correspondence: Kurt V Mikkelsen, Department of Chemistry, University of Copenhagen, Universitetsparken 5, 2100, Copenhagen Ø, Denmark, Email: kmi@chem.ku.dk
Today’s need for renewable energy combined with modern societies' reliability on on-demand power leads us to find solutions that can store excess or produce directly to storage for later use. A MOlecular Solar Thermal (MOST) based on norbornadiene/quadricyclane(NBD/QC) does the latter with an isomeric photoswitching molecule pair. The theoretical studies of molecular solar thermals (MOST) provide a needed understanding of potential synthetic candidates. We have investigated an array of more complex solvation models for the norbornadiene/quadricycle (NBD/QC) photoswitch and the impacts of the models on the first absorption energy. Our results have been obtained with various density functional theoretical methods and basis sets.
In the shift towards renewable energy sources, harnessing solar energy emerges as a pivotal focus due to the sun’s abundance as an untapped energy reservoir [1,2]. Molecular photoswitches offer a promising, cost-effective avenue for potentially storing solar energy over extended periods compared to conventional photovoltaic solutions [3-6]. Through molecular solar thermal energy storage (MOST) systems, employing photoswitchable parent molecules, a chemical transformation occurs upon exposure to sunlight, transitioning into a metastable, higher energy state [7,8]. The stored solar energy is subsequently released as heat when the metastable state reverts to the parent form. Catalytic control, using homogeneous and heterogeneous catalysts, allows for the on-demand release of energy.
However, identifying the optimal photoswitch for MOST applications necessitates optimization across various parameters. The system must absorb light in the UV-visible range corresponding to the solar spectrum in its parent form, while the metastable form should exhibit minimal absorption. Additional criteria include high stability against photodegradation and reactions with species like singlet oxygen, a high quantum yield of photoisomerization, and low molecular mass for increased energy density [9,10].
This study focuses on the norbornadiene/quadricyclane (NBD/QC) couple, a photochromic molecular system, and explores the potential influence of solvent molecules and metallic nanoparticles (NPs) on its thermochemical properties for enhanced MOST applications. Earlier findings suggested that solvent molecules and NPs could significantly reduce the thermal back reaction barrier of related couples without compromising solar energy storage [11]. Additionally, solvent molecules and NPs were found to enhance one and two-photon absorption, induce redshifts in absorption, and alter electric properties [12-14]. Consequently, coupling such solvent molecules and NPs with the NBD/QC system could catalyze the thermal back reaction and modify optical properties for efficient solar energy storage.
The study focuses on an NBD/QC system known for its effective performance in MOST applications utilizing both catalytic and electrochemical activation [15-17]. The primary question addressed is whether the thermal back reaction can be promoted without compromising energy storage capabilities using NPs. The key inquiries include changes in storage energy and thermal back reaction barriers upon physisorption onto different NP surfaces, the dependency on NP type, and the impact of distance and relative orientation between NBD/QC systems and the solvent molecules or NPs.
Hybrid quantum mechanical/molecular mechanical (QM/MM) calculations are employed to model the NBD/QC system and the solvent molecules using quantum mechanics, while the NP is modeled using molecular mechanics. Interaction between the subsystems is determined through polarization dynamics, providing an atomistic description of the NPs. The study employs various orientations of NBD/QC relative to the solvent molecules and the NP to investigate the influence of the NP and the solvent molecules. The investigation aims to contribute insights into leveraging metallic NPs to enhance the applicability of the NBD/QC system in MOST, shedding light on the complex interplay between molecular photoswitches and nanoparticle surfaces.
We [18] have shown that M06-2X and ω B97XD with def2-TZVP or 6-311+G(d) produce good results for an NBD/QC [19] system in a vacuum and a continuum solvation model. To take our understanding further we wish to model this system in more detail to check the accuracy of our earlier work and to investigate how the absorption profile of the systems changes as a result of a variance in solvation and modeling parameters. Our aim is to gain knowledge of how NBD/QC’s properties evolve through increasing solvation modeling complexity, specifically the first absorption peak as well as the intensity of its oscillator strength, which we deem to be good representations of the influence of the models.
We have done this for both the carboxylate and neutral species shown in Figure 1, denoted as NBD(1)/QC(1) and NBD(2)/QC(2) respectively.
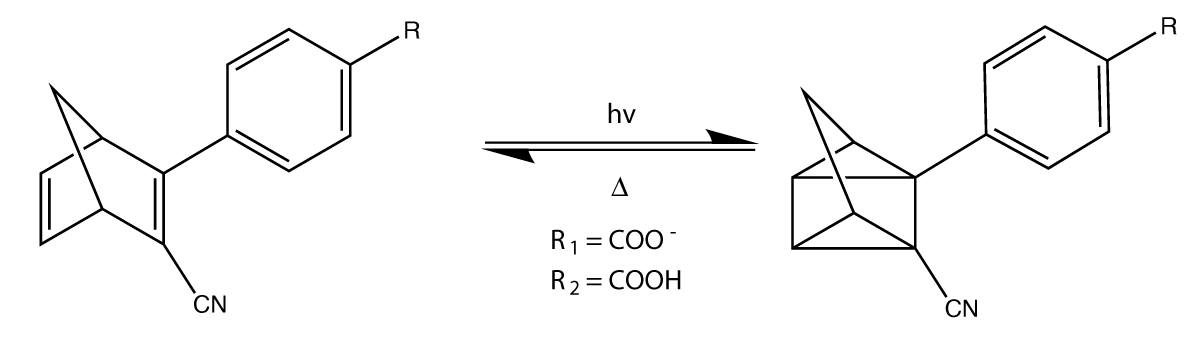
Figure 1: The investigated NBD/QC photoswitch and its reaction path.
Computational details
Calculations were performed using Density Function Theory (DFT) at the level of CAMB3LYP [20,21], M06-2X [22], MN15 [23], SOGGA11-X [24], ω B97XD [25] with the basis sets 6311+G(d), 6-311++G(d,p) [26-33] and def2-TZVP [34,35] levels of theory using Gaussian16 [36]. The calculations were performed with the presence of methanol (∈st = 32.613, ∈op = 1.766) as both an explicit molecule and the combination of the explicit molecule and the IEF-PCM method [37]. Thereby, we implicitly model the outer solvent as a dielectric medium. We compare the results with UV-Vis spectra in toluene(∈st = 2.374, ∈op = 12.238). The two model abstractions, the supermolecular model (SMM) and the semi-continue model (SCM) are outlined below along with the vacuum and the continuum model (CM) (by Bieske, et al. [38]) Figure 2.
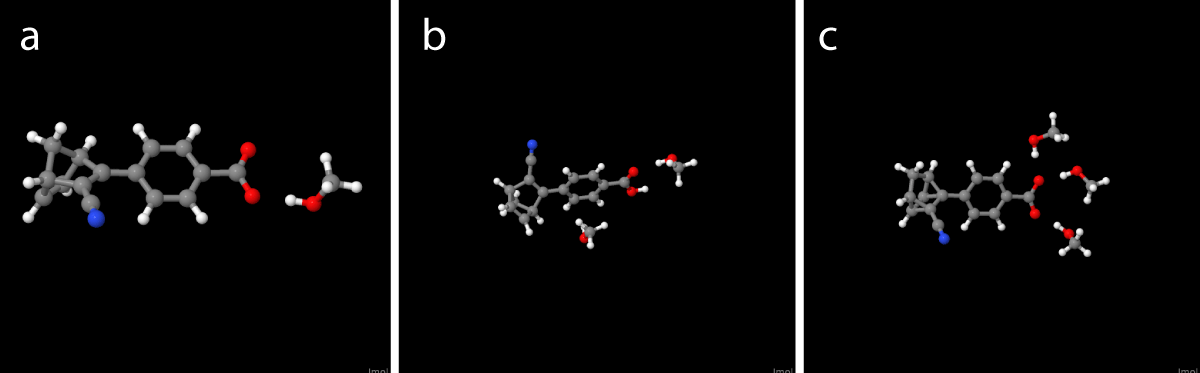
Figure 2: The investigated explicit solvent molecule systems with one, two and three explicit solvent molecules as both charged and uncharged species. The one explicit solvent molecule system (Figure 2(a)) have been investigated with varying degrees of theory, while both the two and three (Figures 2(b) and 2(c)) molecule solvent systems have been performed with DFT/CAM-B3LYP/6-311+g(d) level of theory.
Model abstractions
Vac - Calculations performed in a vacuum
CM - Continuum Model, calculations performed an IEF-PCM solvent shell
SSM - Super Molecular Model, calculations performed in a vacuum with one explicit solvent molecule calculated
SCM - Semi Continuum Model, the investigated molecule and one explicit solvent molecule inside of an IEF-PCM solvent shell.
Time-dependent DFT (TD-DFT) calculations were performed in order to obtain excitation energies and oscillator strengths. With these, UV-Vis spectra were simulated under the assumption of Gaussian band shape with a standard deviation of σ = 0.4, with fifteen electronic excitation states calculated per molecular configuration.
We have in addition to our solvation investigation studied the interaction between our molecular systems and a hemispherical gold nanoparticle. This has been done using DFT and the M06-2X/6-311+G(d) level of theory for the reference state and DFT with the CAMB3LYP/aug-cc-pVDZ [39,40] level for the interaction. This has been performed in Gaussian16 and Dalton20, respectively.
The alignments of the systems relative to the hemisphere were done as shown in Figure 3 both as pictured with methanol, but calculations without methanol have also been performed. We have used ChemDraw and Jmol [41] to generate our molecular structures, with the latter being used to illustrate charge distribution within the molecules.
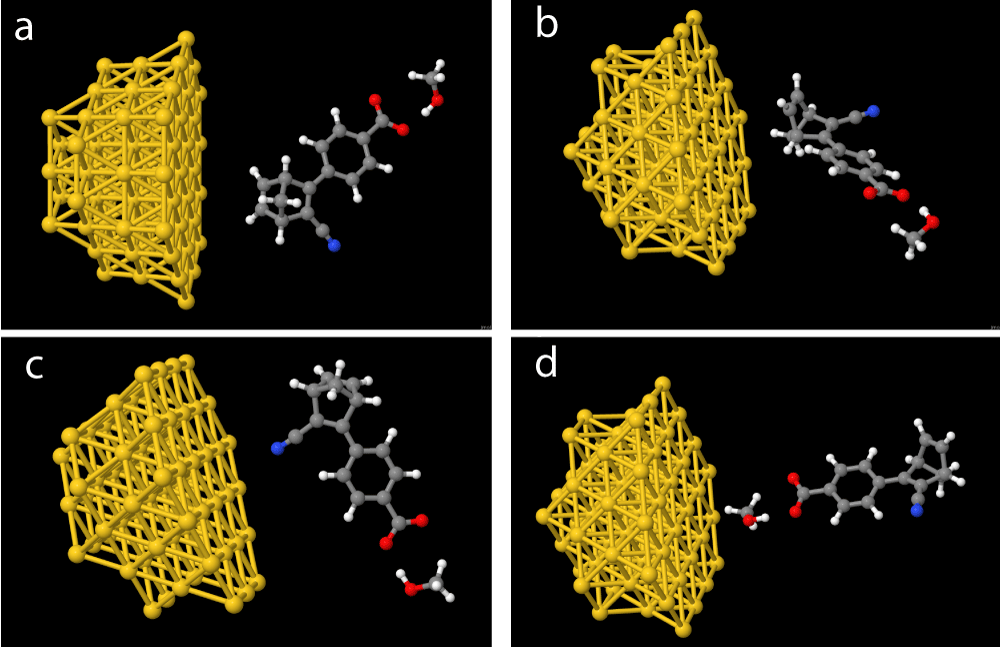
Figure 3: Investigated molecular-nanoparticle configurations with the molecular systems alignment relatively to the gold hemisphere outlined, (a) side orientation (b) top orientation (c) CN orientation and, (d) COO- orientation.
Solvation environment
We have looked at the effect of adding explicit solvent molecules to NBD carboxylate (compound 1) in a vacuum, by looking at the first absorption energy. This results in a blueshift as shown in Figures 4(a,b), with the incrementally shift decreasing with additional solvent molecules, as shown in Figure 4c. This trend of blueshift as a consequence of the introduction of an explicit solvent molecule is amplified for NBD(2), shown in Figure 5, however, further blueshift with additional solvent molecules is not occurring for NBD(2), where the absorption consistently lies at ≈ 301 nm, see Table 1.
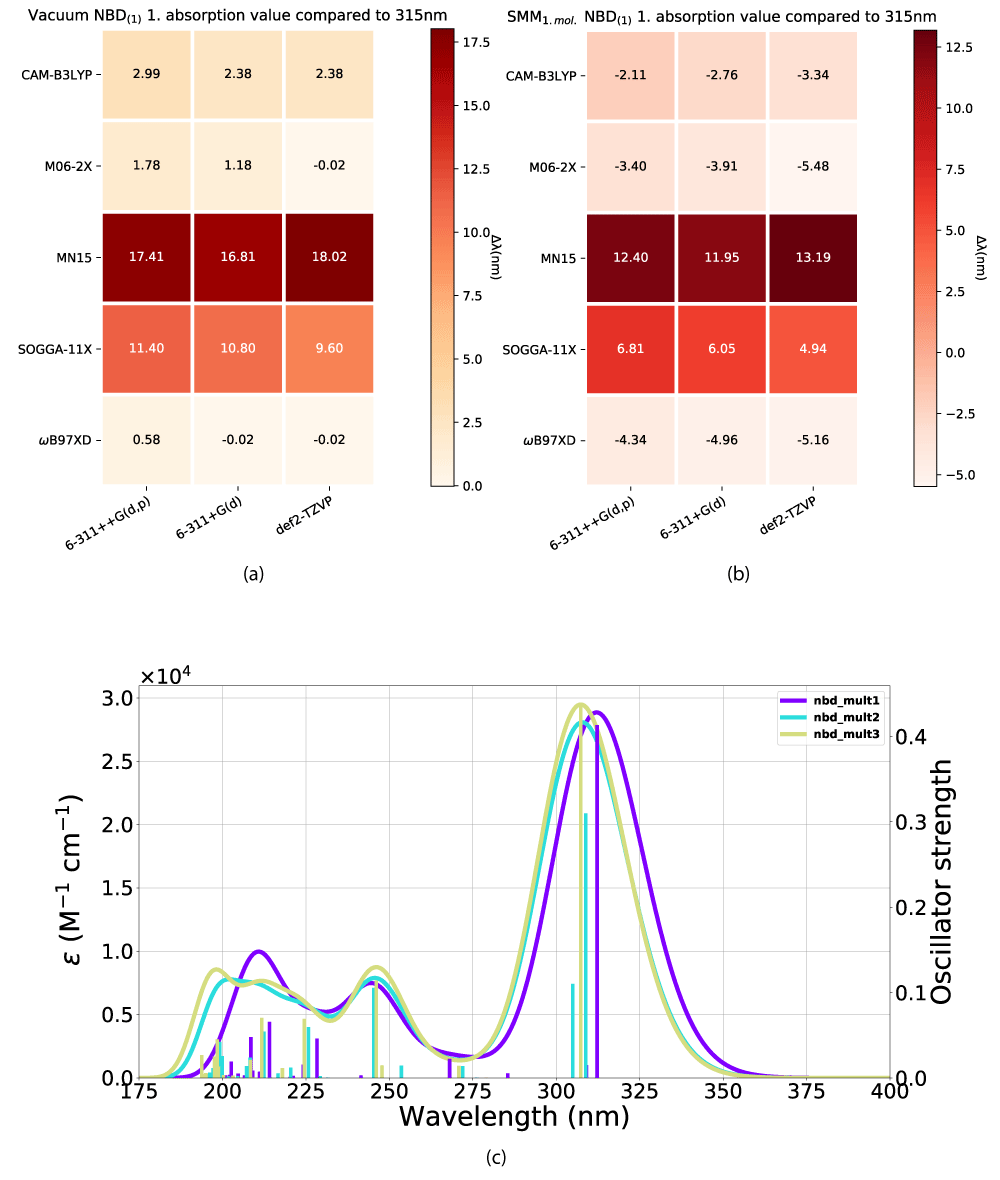
Figure 4: Effect from the addition of explicit solvent molecules to NBD carboxylate (compound 1) in vacuum. Vacuum ((a)) and experimental data taken from Bieske, et al. [38], (b) SCM model. (c) The x-axis is the basis set and the y-axis is the DFT method.
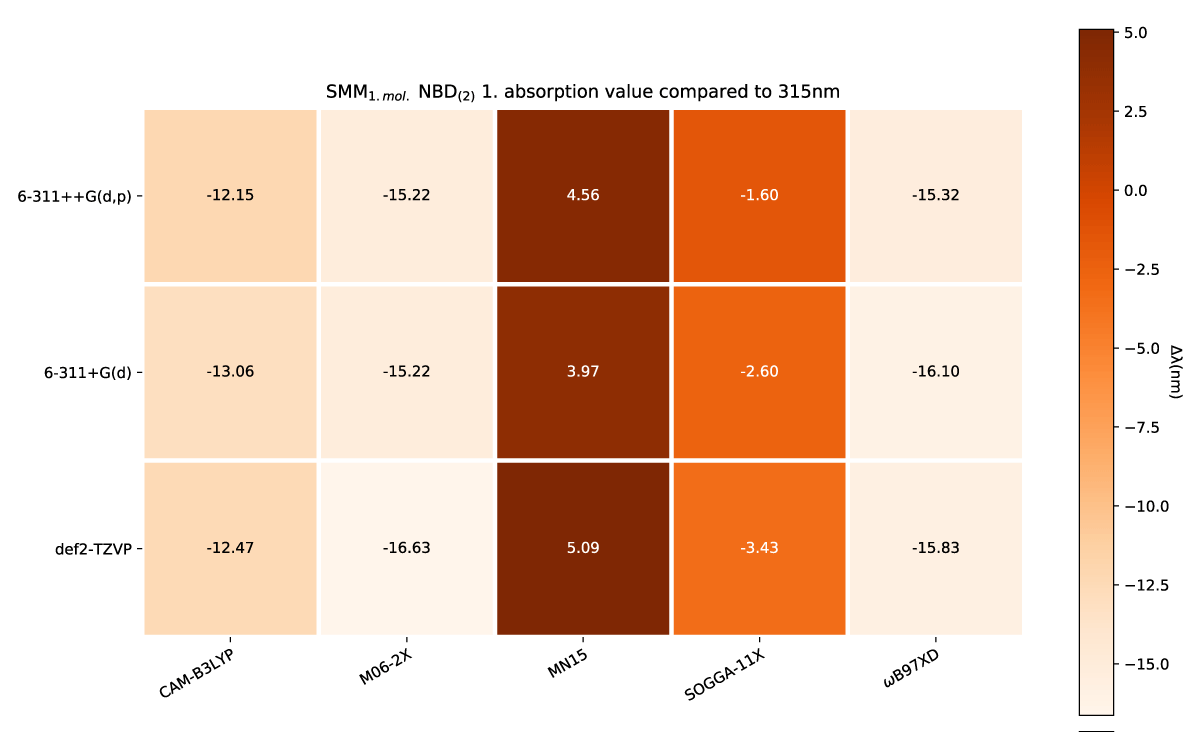
Figure 5: First absorption values of SMM model NBD(2) compared to experimental PISA [38] value.
| Table 1: First absorption peak for both NBD and QC calculated with CAM-B3LYP/6-311+G(d) level of theory Vac and solCM data point's by Bieske, et al. [38]. | |||||||
| 1/nm | lcalc | lmax | lonset | lcalc | lmax | lonset | 1/nm |
| NBD(Vac) | -/31738 | 315 | 355 | -/22038 | - | - | QC(Vac) |
| NBD(sol CM) | -/30338 | 31942/31538 | 37842/35538 | -/23238 | 256 | 296 | QC(solCM) |
| NBD(sol SMM-1) | 301/312 | - | - | 249/237 | - | - | QC(solSMM-1) |
| NBD(sol SCM-1) | 308/305 | - | - | 256/232 | - | - | QC(solSCM-1) |
| NBD(sol SMM-2) | 301/309 | - | - | 249/228 | - | - | QC(solSMM-2) |
| NBD(sol SCM-2) | 311/304 | - | - | 254/235 | - | - | QC(solSCM-2) |
| NBD(solSMM-3) | 303/307 | - | - | 247/224 | - | - | QC(solSMM-3) |
| NBD(solSCM-3) | 309/306 | - | - | 264/239 | - | - | QC(solSCM-3) |
The charged QC(1) conformer also displays an incrementally decreasing blueshift with additional solvent molecules. Our calculations with one explicit solvent molecule, however, are closer to the experimental values than the pure vacuum value calculated by Bieske, et al. [38], as shown in Figure 6a and Table 1. Furthermore, our multi explicit solvent molecule calculations dip below the vacuum value of 220 nm, as displayed in Table 1. The elimination of the molecular charge narrows the gap between our calculated values and the experimental reference value, while the incremental blueshift as a result of additional solvent molecules is almost eliminated, also shown in Table 1.
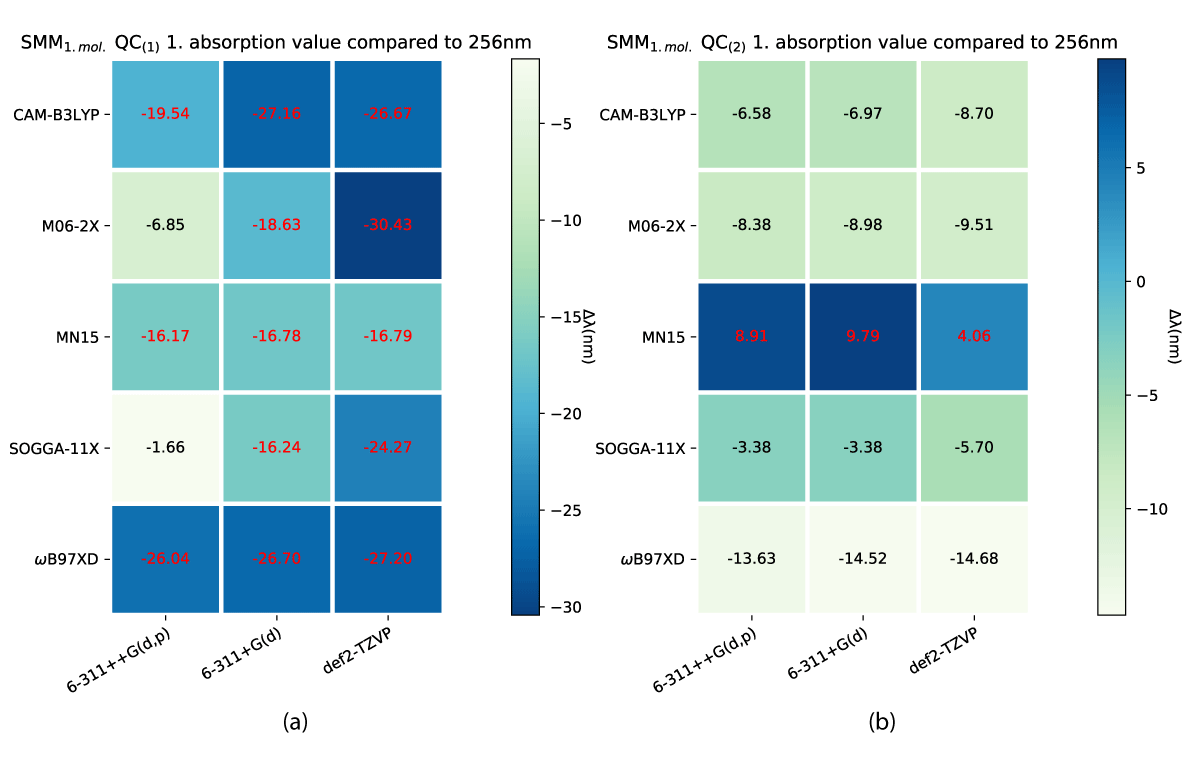
Figure 6: Effect of the addition of explicit solvent molecules to QC carboxylate (compound 1) in vacuum. Vacuum (a) and experimental data taken from Bieske, et al. [38], (b) and SCM model. In (a) and (b) the x-axis is the basis sets and the y-axis is the DFT method.
Looking at the solvated charged case of the CM and SCM models we get a slight redshift of the absorption by including an explicit solvent molecule going from CM to SCM, as shown in Figures 7(a,b), which is carried onwards ever so slightly with additional solvent molecules as shown in Figure 7c.
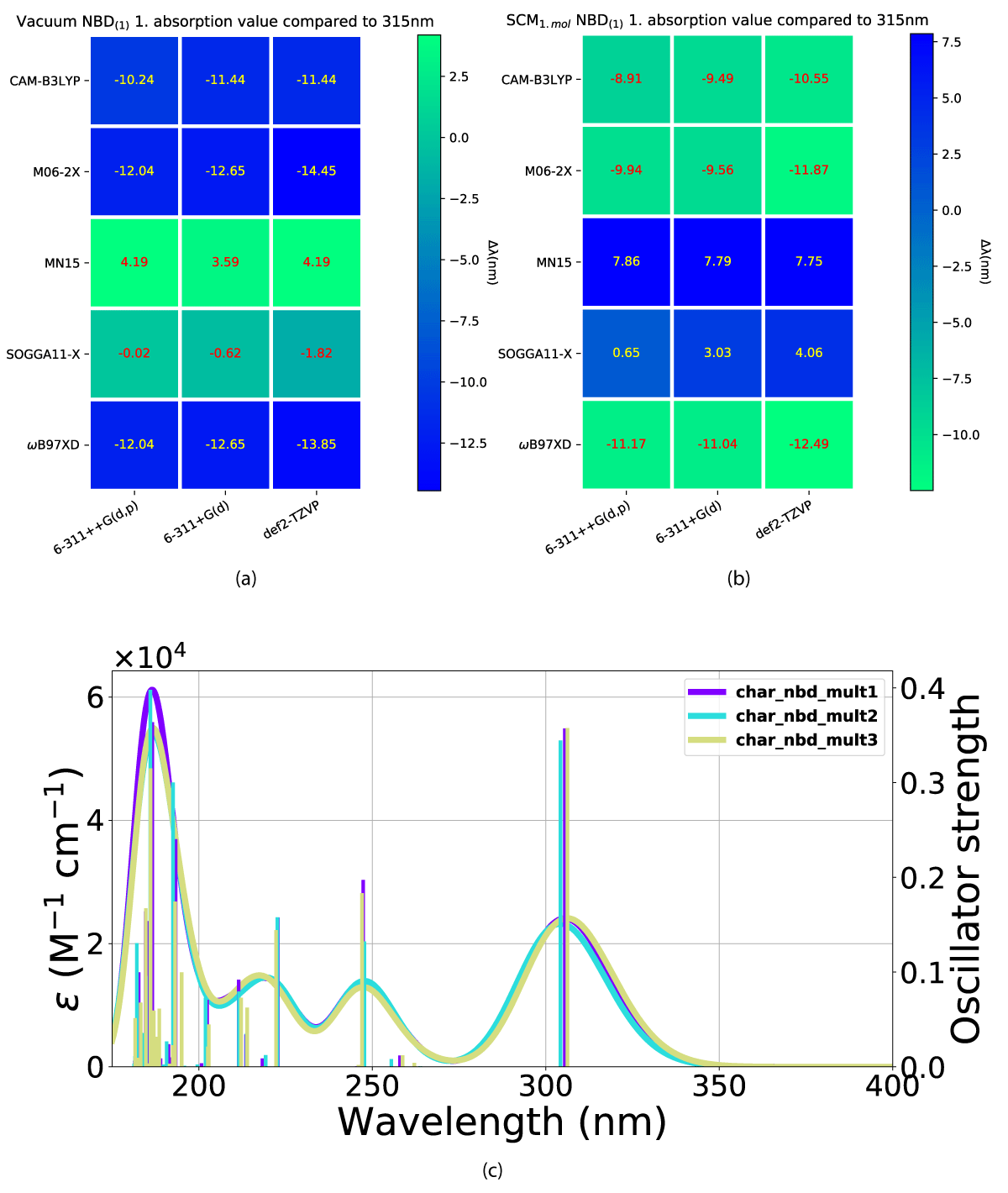
Figure 7: Effect from the addition of explicit solvent molecules to NBD carboxylate (compound 1) in vacuum. Vacuum ((a)) and experimental data taken from Bieske, et al. [38], (b) SCM model. (c).
The effect of a charge shift from 1 to 2 results in a slight further redshift of the first absorption energy, which again is echoed with more explicit solvation molecules, as shown in Figures 8,9, respectively.
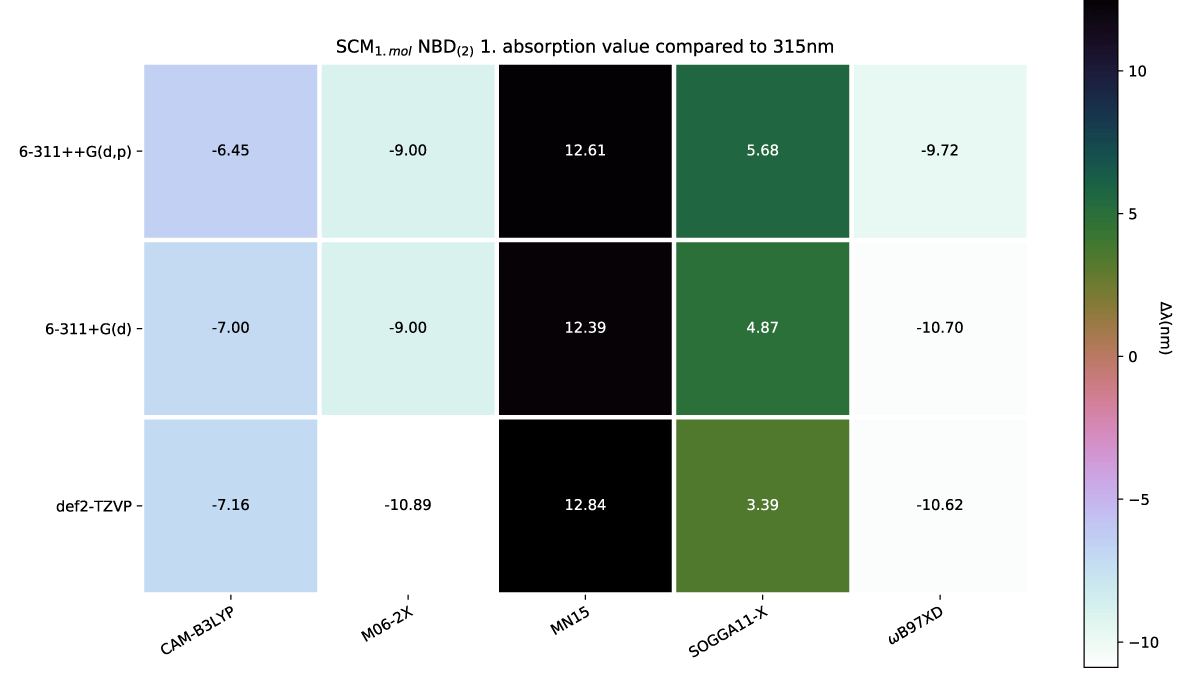
Figure 8: SCM NBD(2) variens from experimental value of 319nm [42].
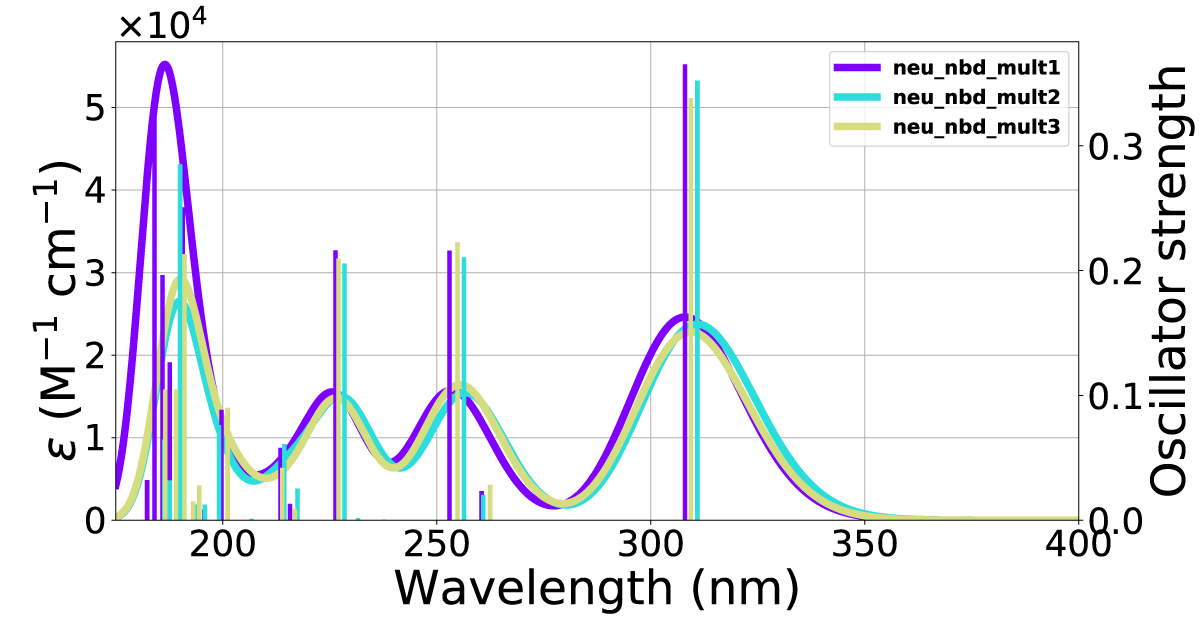
Figure 9: Calculated UV-Vis spectra including 1-3 explicit solvent molecules around NBD(2) at the CAM-B3LYP/6-311+G(d) level of theory.
While both the charged and uncharged NBD conformers (NBD(1) and NBD(2)) do lie inside a narrow margin of error in predicting the first absorption energy, this is not the case for QC. Our results show significant differences between QC(1) and QC(2) which are shown in Figure 10. The smallest divergence for the charged case is the biggest for the uncharged, which furthermore, predicts the experimental value at some of the theory levels, as shown in Figure 10b.
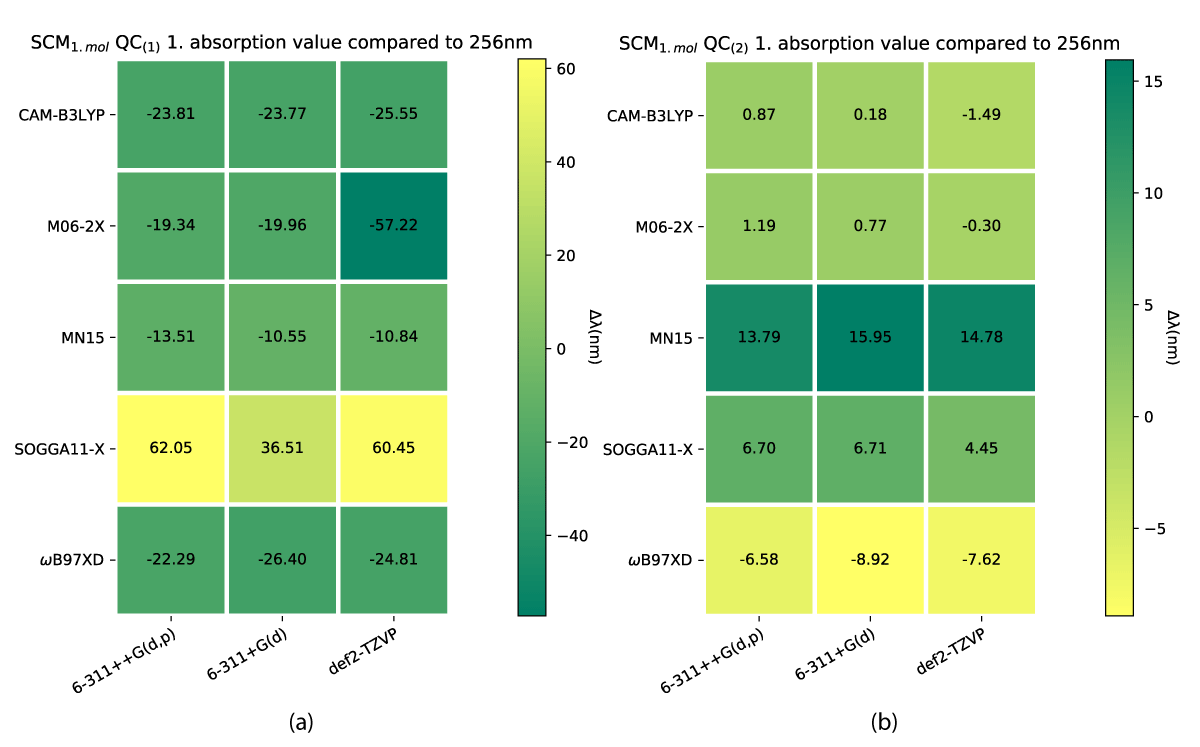
Figure 10: First absorption eneriges for SCM model QC(1) (a) and QC(2) (b) varying from experimental value of 256 nm [38].
This large disparity between charged and uncharged species in first absorption energy have lead us to investigated the uncharged specie in greater detail and compare them as shown in Figures 12,13 and look at the placement of the solvent molecules around the NBD/QC photoswitch. We have in addition compared our results to experimental reference data from UV-Vis experiments [42] performed in toluene, shown in Figure 11, the error range of which is comparable to that of the CM model by Bieske, et al. [38] (Figure 7b) and our SMM model results (Figure 4b).
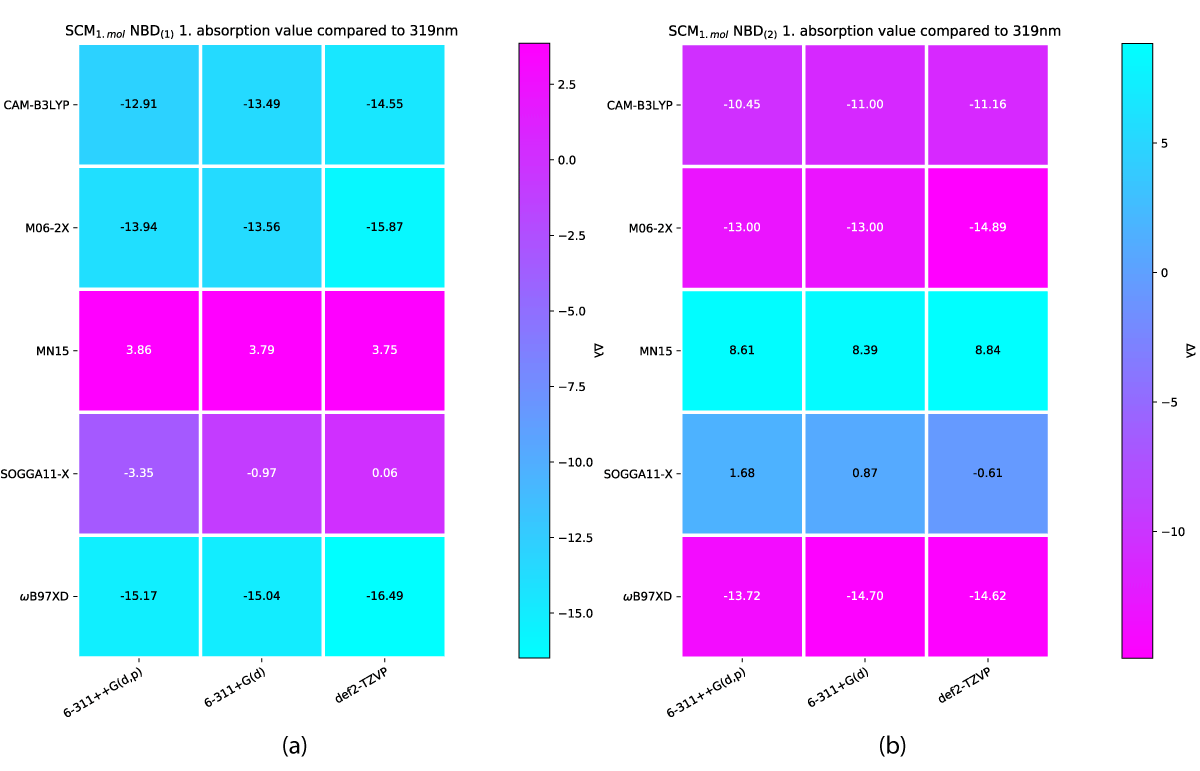
Figure 11: SCM model NBD(1) (a) and NBD(2) (b) compared to first absorption value from UV-Vis42 performed in in toluene.
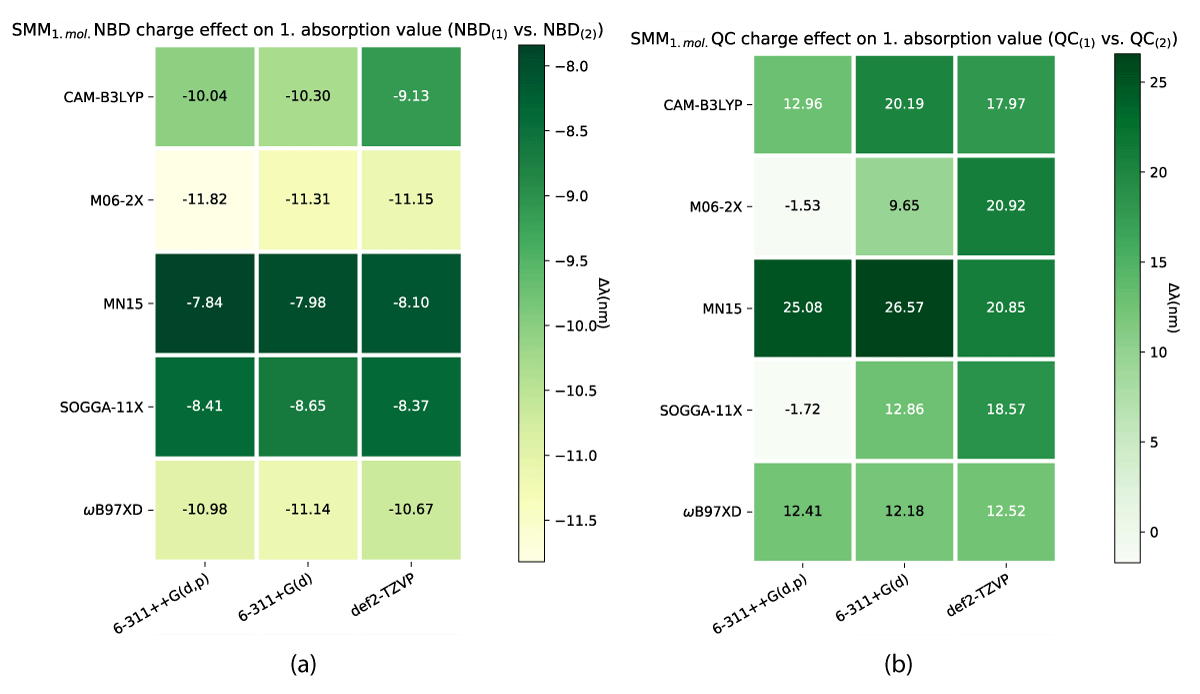
Figure 12: Comparison of first absorption energy between differing molecular charges within the models shown with (a) SMM NBD and (b) SMM QC.
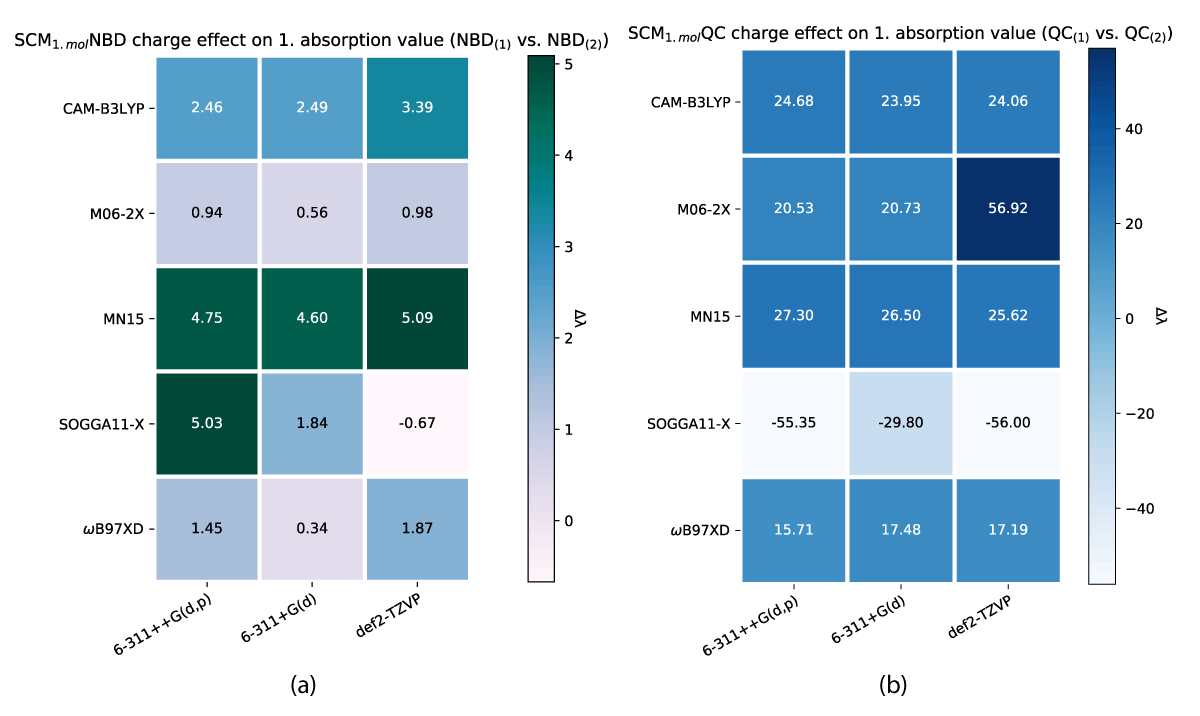
Figure 13: Comparison of first absorption energy between differing molecular charges within the models shown with (a) SCM NBD and (b) SCM QC.
In Table 1 we see that our calculated values for SCM/QC with one explicit solvent molecule have the same absorption value as the experimental GC-MS value by Bieske, et al. [38], i.e. an experimental GC-MS molecule, which should be charged, have an absorption value corresponding to that of a uncharged calculated molecule.
We have, due to this discrepancy, compared the first absorption peaks of NBD and QC in both of our models to find the effects of the charge of the neutral acid group versus the charged carboxylate, which are shown in Figures 12,13. These changes do as shown, affect the QC conformers to a larger extent than in the case of NBD. Along with the impact of more explicit solvent molecules, as shown in Table 1, these do point to a clear trend of the QC conformers being volatile to environment changes, which to us indicates insufficiencies in the level of theory used to model this system.
The comparisons of absorption above (Figures 4(a,b), 5, 6(a,b), 7(a,b), 8, 10 and 11) have been made to non-optimal experimental reference values, as both PISA [38] and UV-Vis [42] values are inadequate for our total model space and as such should be viewed with precaution. We do, however, see the trends as representative, from which we are able to say that increased model complexity benefits the QC conformer while being inconclusive for NBD.
We have done a preliminary investigation on the system to explore these trends further, with increased solvation complexity at the CAM-B3LYP/6-311+G(d) level of theory, shown in Table 1. The spectrum of QC converges to and around the experimental first absorption value from PISA [38], indicating the presence of solvent molecule at detection and a possible proton transfer as our results with neutral NBD (NBD(2)) align the best with said value.
These results illustrate the marginal gains in accuracy of more complex solvent models for the NBD system, but appraisal for QC. This indicates sufficient accuracy afforded with a CM approach in modeling NBD.
Charge effects on first absorption energy
As one might note in Table 1, there are distinct differences in absorption between the charged and uncharged molecules for both NBD and QC, which is why we have looked at the charge effect on the absorption as illustrated in Figures 12, 13.
As described above, a change in the molecular charge results in a shift in absorption. This shift is nonuniform through the models as illustrated in Figures 12,13. As shown the SCM model induces both the largest and smallest shíft in absorption due to changing charge, with NBD being affected negligently and QC experiencing a ≈ 100 nm change in the shift. While the effect is closer between the conformers for the SMM model (Figure 12), QC still has a larger change overall.
Storage energies
We have as part of our investigation calculated the storage energies for both molecular configurations in both of our models, the results of which are shown in Table 2.
| Table 2: Calculated storage energies of the charged molecule in the investigated solvation models, displayed in kJ/mol. | |||
| Gray SSM(kJ/mol) | 6-311++G(d,p) | 6-311+G(d) | def2-TZVP |
| Charged species | |||
| CAM-B3LYP | 107.3 | 108.2 | 104.0 |
| M06-2X | 86.2 | 88.6 | 86.2 |
| MN15 | 93.9 | 95.4 | 91.7 |
| SOGGA11-X | 89.2 | 88.7 | 87.1 |
| wB97XD | 95.2 | 94.5 | 93.1 |
| Neutral species | |||
| CAM-B3LYP | 109.6 | 112.3 | 106.6 |
| M06-2X | 72.9 | 145.3 | 71.0 |
| MN15 | 90.6 | 90.0 | 87.0 |
| SOGGA11-X | 71.9 | -13.0 | 70.9 |
| wB97XD | 80.9 | 79.8 | 76.4 |
| SCM(kJ/mol) | 6-311++G(d,p) | 6-311+G(d) | def2-TZVP |
| Charged species | |||
| CAM-B3LYP | 94.2 | 123.4 | 95.2 |
| M06-2X | 93.4 | 94.5 | 106.3 |
| MN15 | 109.5 | 102.6 | 76.7 |
| SOGGA11-X | 83.5 | 95.8 | 73.9 |
| wB97XD | 98.5 | 102.8 | 80.2 |
| Neutral species | |||
| CAM-B3LYP | 118.9 | 122.5 | 119.0 |
| M06-2X | 76.8 | 149.7 | 73.7 |
| MN15 | 99.7 | 103.9 | 101.6 |
| SOGGA11-X | 76.171 | -11.1 | 74.2 |
| wB97XD | 80.5 | 83.4 | 78.6 |
As shown in Table 2 the shift between neutral and charged molecules relates to a change in the storage energies, specifically the SOGGA11-X/6-311+G(d) level of theory, which inverts the preferred molecular configuration in both models.
Generally, our methods show better consistency with the change of model than change of charge, CAM-B3LYP being the most consistent with the charge difference.
Our results show unexpected changes in storage energies as a result of different basis sets, especially the 6-311+G(d) basis set exhibits large differences in energy compared to 6311++G(d,p) and def2-TZVP when calculated with the same method.
Micro-solvation structure
The different charge configurations (1 and 2) of the NBD/QC pair affect how the explicit solvent molecules are placed around them as shown in Figure 14. This might be one of the contributing factors to the change in absorption and energies observed as a result of the charge shift, the most radical of these being the SOAGGA11-X functional.
The lack of charge allows for the solvent interaction with a point differing from the carboxylate/acid group. As shown in Figure 14d the interaction between the charge and the dipole moments of the solvent molecule is strong compared to that of the neutral species (Figure 14c) and while the solvent molecules do move away from the point charge of the carboxylate group (1), they have less tendency to do so with the neutral (2) molecule.
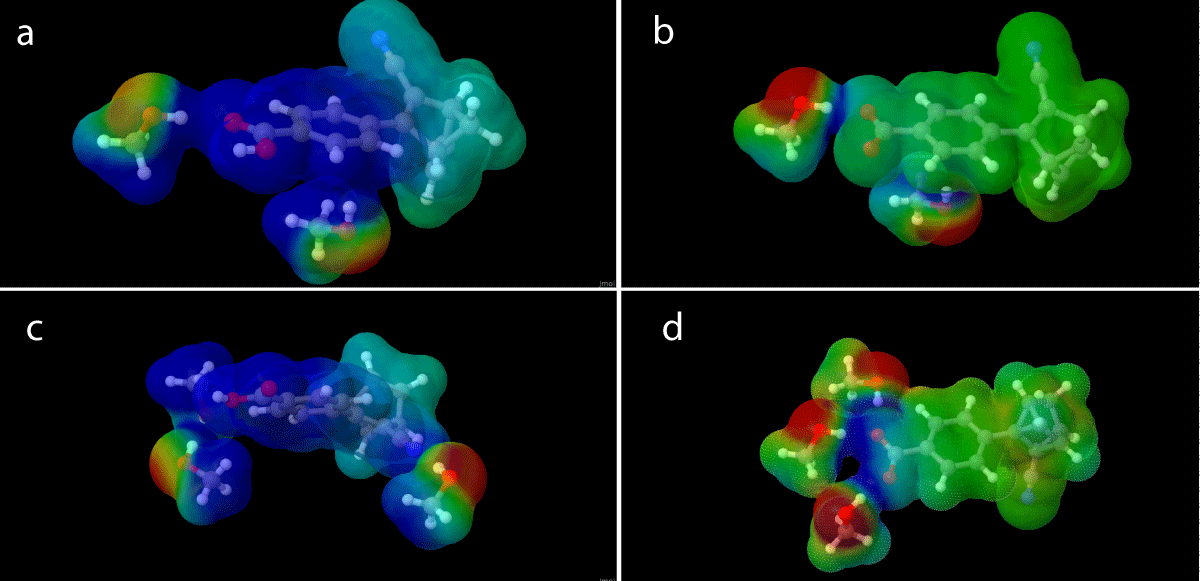
Figure 14: Micro-solvation structure of the charged QC (a) and neutral (b) QC with two explicit solvation molecules, along with micro-solvation structures with three explicit solvation molecules for charged QC (c) and neutral NBD (d) molecules.
Gold nanoparticle interaction
We have looked at the interaction of our system with a gold nanoparticle both with and without an explicit solvent molecule at different distances between the gold nanoparticle and the molecular system. We have compared four different systems to nanoparticle configurations for both the NBD and QC conformer, shown in Figures 15,16, respectively.
As shown in these figures (Figures 15,16) the interactions result in a minor blueshift of all NBD(1) alignment but that of Figure 3d which is shown in Figure 15d, thereby mirroring the trends exhibited by the addition of explicit solvent molecule to NBD/QC.
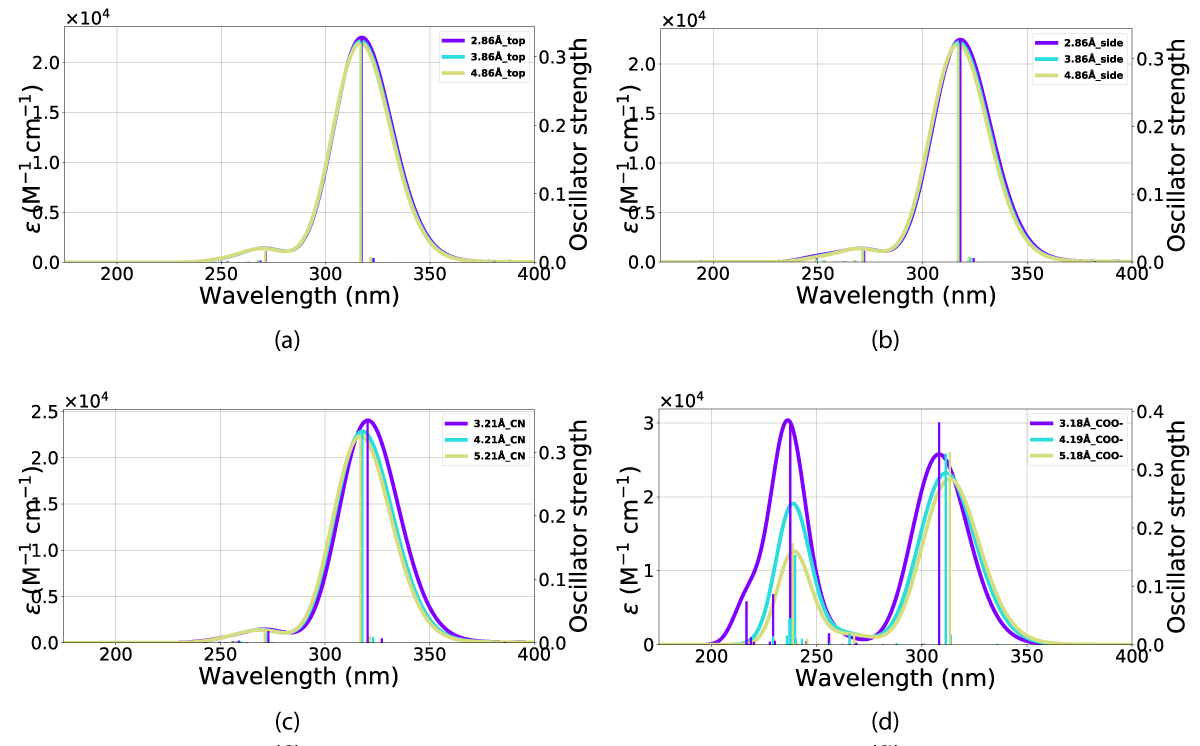
Figure 15: UV-VIS spectra for NBD in interaction with a gold nano-particle at various distances (in Ångstrom) and orientations. The x-axis is the wavelength and the y-axis is the intensity. The orientations for the respective UV-VIS spectra can be seen in the following figures: (a) Figure. 3(b), (b) Figure. 3(a), (c) Figure. 3(c) and (d) Figure 3(d) orientations.
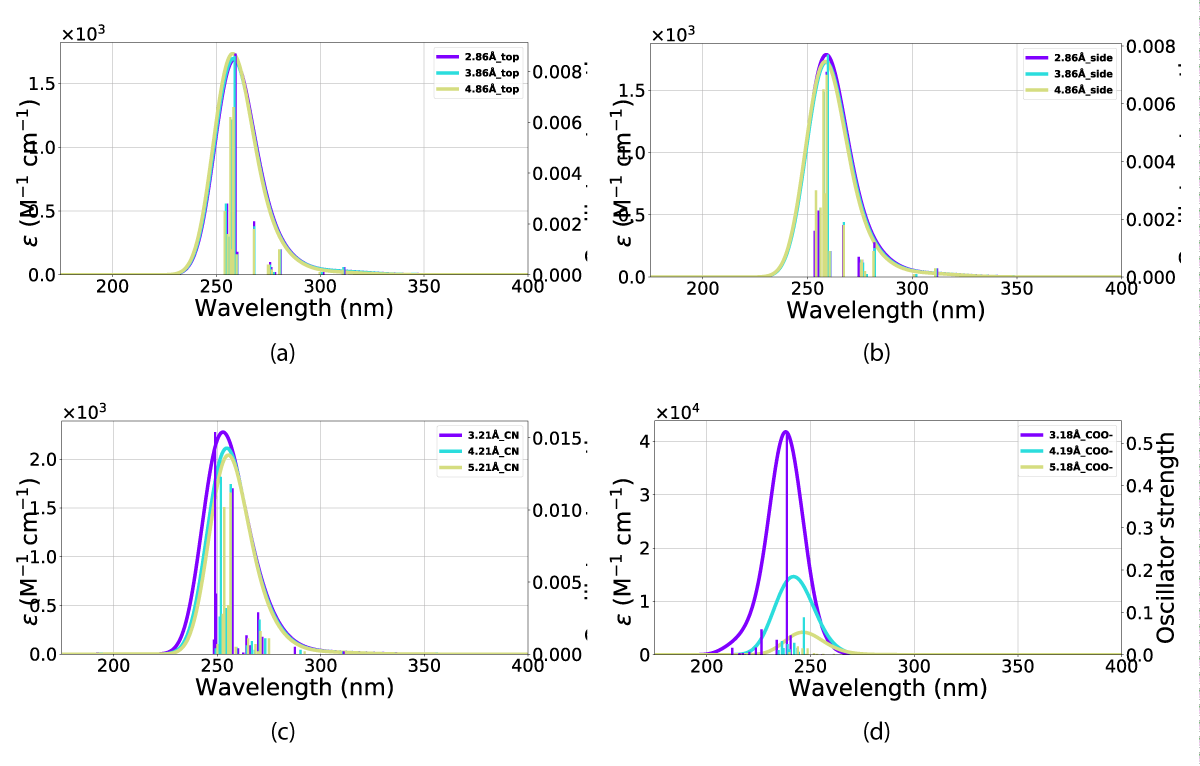
Figure 16: UV-VIS spectra for QC in interaction with a gold nano-particle at various distances (in Ångstrom) and orientations, (a) top orientation in various distances (b) side orientation (c) CN orientation and, (d) COO- orientation. The x-axis is the wavelength and the y-axis is the intensity.
The effect on the QC(1) conformer is an increased absorption oscillator strength, this effect is most visible for the COO alignment (Figure 3d) shown in Figure 16d. However, this is not true for the top alignment (Figure 3b), which exhibits a small decrease in the absorption intensity (Figure 16a) as a result of the interactions with the nanoparticle.
The effects of including an explicit solvent molecule, shown with NBD(1) in Figure 17, do confer a small but consistent blueshift of the absorption. This effect results in a move of the peak in Figure 17c from 327 nm - 324 nm absorption peak without an explicit solvent molecule towards 318 nm - 314 nm with one solvent molecule, these effects are shown in Table 3.
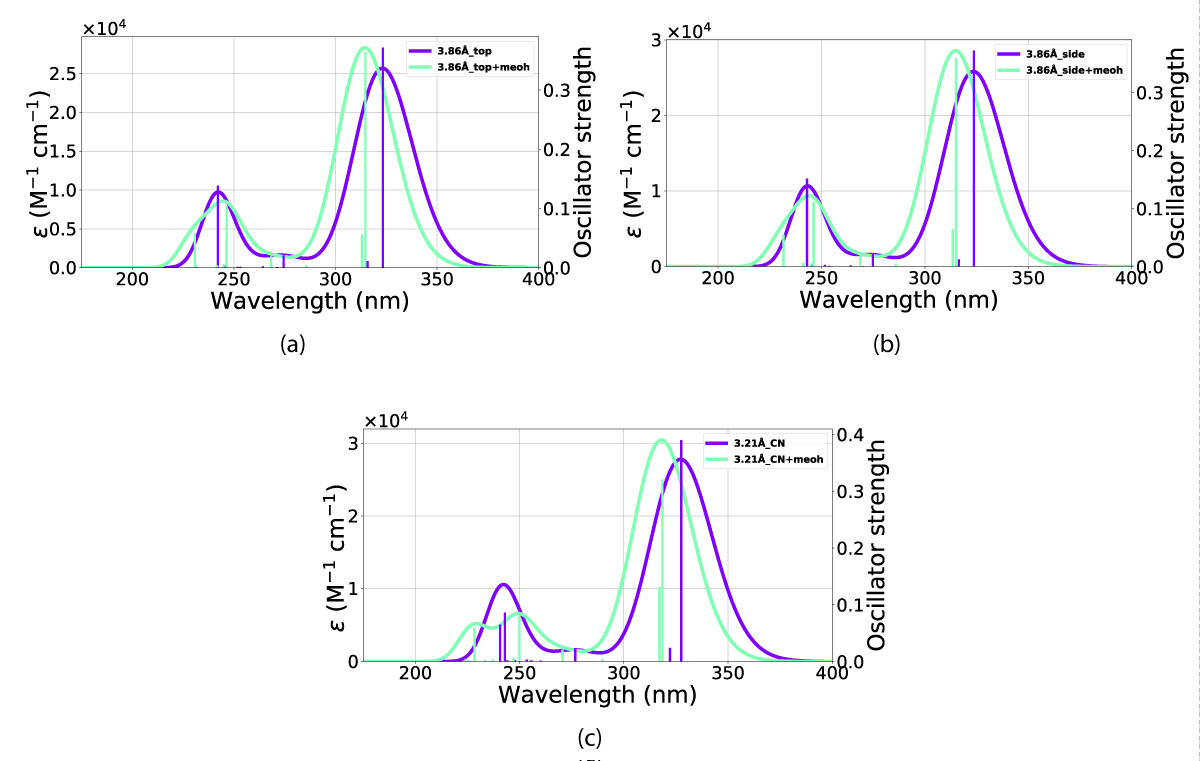
Figure 17: Comparison of UV-Vis spectra between inclusion and exclusion of an explicit solvent molecule in the (a) top, (b) side and (c) CN orientations. The x-axis is the wavelength and the y-axis is the intensity.
| Table 3: Peak absorption with and without explicit solvent molecule included in reference state. | ||
| Orient. | With(nm) | Without(nm) |
| NBDCN (3.21Å) | 318 | 327 |
| NBDCN (4.21Å) | 315 | 324 |
| NBDCN (5.21Å) | 314 | 323 |
| NBDside (2.86Å) | 315 | 324 |
| NBDside (3.86Å) | 315 | 323 |
| NBDside (4.86Å) | 314 | 323 |
| NBDtop (2.86Å) | 315 | 323 |
| NBDtop (3.86Å) | 314 | 323 |
| NBDtop (4.86Å) | 314 | 323 |
| NBDCOO | 311(3.86Å) | 316(3.18Å) |
| NBDCOO | 312(4.86Å) | 318(4.19Å) |
| NBDCOO | 313(5.86Å) | 320(5.18Å) |
The result of including an explicit solvent molecule into the molecule (NBD/QC) nano-particle interaction is a consistent blueshift in absorption. The absorption of the nanoparticle and molecular system does, however, lie at a longer wavelength than the molecule alone, resulting in overlaps between the nanoparticle, molecule, and explicit solvent molecule system with that of NBD in vacuum (calculations and PISA) and solvation (UV-Vis).
This overall blueshift is dependent on the relative orientation between the molecular system and the nanoparticle. As shown in both Figures 15,16 the interactions of the gold particle and system (NBD/QC) only convey a small distortion of the absorption. This effect is mostly dependent on the substituent aligned with the particle. However, the distance between the two also plays a role in intensity, which is the most pronounced with the carboxylate group aligned against the nanoparticle Figure 3d and that is illustrated in Figure 15d and Figure 16d. Illustrated in Figure 16 is the fact that the QC-carboxylate species do not absorb in the visual spectrum.
The results of including an explicit solvent molecule into the molecule (NBD/QC) nanoparticle interaction are consistent blueshifts in absorption. The absorption of the nanoparticle and molecular system does, however, lie at a longer wavelength than the molecule alone, resulting in overlaps between the nanoparticle, molecule, and explicit solvent molecule system with that of NBD in vacuum (calculations and PISA) and solvation (UV-Vis).
This overall blueshift is dependent on the orientation between the molecule and particle, as shown in both Figures 15,16, the interaction of the gold particle and system (NBD/QC) only conveys a small distortion of the absorption. This effect is mostly dependent on the substituent aligned with the particle, however, the distance between the two bodies also plays a role in the intensity, which is the most pronounced with the carboxylate group aligned against the particle Figure 3d illustrated in Figures 15d, 16d. Illustrated in Figure 16 is the fact that the QC-carboxylate species do not absorb in the visual spectrum which is a significant advantage.
As we have outlined the increased model complexity results in a small and incrementally decreasing blueshift of the first absorption value of NBD. QC shows a beneficial response from the investigated models, however, since QC absorbs below 300 nm its accurate modeling is of lesser concern.
We have shown that the previously applied model results in a sufficient level of accuracy in the modeling of NBD and increased model complexity does not confer perceptible improvements in this matter.
This study has investigated the environmental effects on the norbornadiene-quadricyclane (NBD/QC) photoswitch, a crucial component in molecular solar thermal energy storage (MOST). The research addressed the need for a comprehensive understanding of molecular candidates in MOST, bridging the gap between theoretical models and the physical reality of the systems under investigation.
The primary goal of this research was to enhance our understanding of the NBD/QC photoswitch’s properties, particularly focusing on the first absorption peak and oscillator strength under varying solvation and modeling parameters. By employing advanced computational methods, we were able to refine the accuracy of previous models and unravel how the absorption profile evolves with the increasing complexity of the solvation.
The investigation involved the use of density function theory (DFT) calculations at different levels, including CAM-B3LYP, M06-2X, MN15, SOGGA11-X, and B97XD, using various basis sets. Explicit solvent molecules and continuum solvation models were considered, and time-dependent DFT calculations were performed to obtain excitation energies and oscillator strengths. The study explored different molecular-nanoparticle configurations and their orientations relative to a gold hemisphere. Both charged and uncharged species (NBD(1)/QC (1) and NBD(2)/QC(2)) are investigated.
The addition of explicit solvent molecules to NBD carboxylate in a vacuum results in a blueshift of the first absorption energy. The impact is more pronounced for QC conformers, especially in charged configurations. Solvation models and the number of explicit solvent molecules influence the absorption profile, emphasizing the sensitivity of QC conformers to environmental changes. A distinct difference in absorption between charged and uncharged molecules was observed for both NBD and QC. Charge shifts lead to non-uniform shifts in absorption, with QC conformers showing larger deviations.
Storage energies were calculated for both charged and neutral species in different solvation models, revealing unexpected changes in energy as a result of different methods. CAM-B3LYP consistently exhibited better consistency with charge differences. The micro-solvation structure of charged and neutral molecules with explicit solvent molecules was analyzed. The charge configurations affected the placement of solvent molecules around the molecules, contributing to the observed changes in absorption and energies. The interactions of the system with a gold nanoparticle were explored, considering varying distances between the bodies. Four different configurations are compared for both NBD and QC conformers.
In summary, this research showed intricate details of the NBD/QC photoswitch, shedding light on its behavior under diverse environmental conditions and providing valuable insights for the development of molecular solar thermal energy storage systems. We are of the belief that the level of theory used (DFT/CAM-B3LYP/6-311+G(d)) is insufficient in the description of QC’s absorption behavior or that there were unknown parameters in reference experiment [38]. The alignments of our calculations with the experimental data (Table 1) and the further development of the relation of absorption to a number of explicit solvent molecules are also shown in Table 1. We hope that our results will lead to further experimental investigations in order to validate the results presented here.
- Service RF. Solar energy. Is it time to shoot for the sun? Science. 2005 Jul 22;309(5734):548-51. doi: 10.1126/science.309.5734.548. PMID: 16040683.
- Lewis NS, Nocera DG. Powering the planet: chemical challenges in solar energy utilization. Proc Natl Acad Sci U S A. 2006 Oct 24;103(43):15729-35. doi: 10.1073/pnas.0603395103. Epub 2006 Oct 16. Erratum in: Proc Natl Acad Sci U S A. 2007 Dec 11;104(50):20142. PMID: 17043226; PMCID: PMC1635072.
- Kucharski TJ, Tian Y, Akbulatov S, Boulatov R. Chemical solutions for the closed-cycle storage of solar energy. Energy Environ Sci. 2011; 4: 4449–4472.
- Moth-Poulsen K, Ćoso D, Börjesson K, Vinokurov N, Meier SK, Majumdar A, Vollhardt KPC, Segalman RA. Molecular solar thermal (MOST) energy storage and release system. Energy Environ Sci. 2012; 5: 8534–8537.
- Lennartson A, Roffey A, Moth-Poulsen K. Designing photoswitches for molecular solar thermal energy storage. Tetrahedron Lett. 2015; 56: 1457–1465.
- Wang Z, Losantos R, Sampedro D, Morikawa Ma, Börjesson K, Kimizuka N, Moth-Poulsen KJ. Demonstration of an azobenzene derivative based solar thermal energy storage system. Mater Chem A. 2019; 7: 15042–15047.
- Kucharski TJ, Ferralis N, Kolpak AM, Zheng JO, Nocera DG, Grossman JC. Templated assembly of photoswitches significantly increases the energy-storage capacity of solar thermal fuels. Nat Chem. 2014 May;6(5):441-7. doi: 10.1038/nchem.1918. Epub 2014 Apr 13. PMID: 24755597.
- Yoshida Zi. New molecular energy storage systems. J Photo Chem. 1985; 29: 27–40.
- Dreos A, Börjesson K, Wang Z, Roffey A, Norwood Z, Kushnir D, Moth-Poulsen K. Exploring the potential of a hybrid device combining solar water heating and molecular solar thermal energy storage. Energy Environ Sci. 2017; 10: 728–734.
- Quant M, Lennartson A, Dreos A, Kuisma M, Erhart P, Börjesson K, Moth-Poulsen K. Low Molecular Weight Norbornadiene Derivatives for Molecular Solar-Thermal Energy Storage. Chemistry. 2016 Sep 5;22(37):13265-74. doi: 10.1002/chem.201602530. Epub 2016 Aug 5. PMID: 27492997; PMCID: PMC5096010.
- Hillers-Bendtsen AE, Kjeldal FØ, Mikkelsen KV. Molecular solar thermal energy storage properties of photochromic molecules physisorbed onto nanoparticles. Chem Phys Lett. 2019; 733: 136661.
- Hillers-Bendtsen AE, Hansen MH, Mikkelsen KV. The influence of nanoparticles on the excitation energies of the photochromic dihydroazulene/vinylheptafulvene system. Phys Chem Chem Phys. 2019.
- Hillers-Bendtsen AE, Mikkelsen KV. The influence of gold nanoparticles on the two photon absorption of photochromic molecular systems. Phys Chem Chem Phys. 2019; 21: 18577–18588.
- Boye IMI , Hansen MH , Mikkelsen KV . The influence of nanoparticles on the polarizabilities and hyperpolarizabilities of photochromic molecules. Phys Chem Chem Phys. 2018 Sep 19;20(36):23320-23327. doi: 10.1039/c8cp03645d. PMID: 30175339.
- Luchs T, Lorenz P, Hirsch A. Efficient Cyclization of the Norbornadiene-Quadricyclane Interconversion Mediated by a Magnetic [Fe3O4−CoSalphen] Nanoparticle Catalyst. Chem Photo Chem. 2020; 4: 52–58.
- Wang Z, Roffey A, Losantos R, Lennartson A, Jevric M, Petersen AU, Quant M, Dreos A, Wen X, Sampedro D. Macroscopic heat release in a molecular solar thermal energy storage system. Energy Environ Sci. 2019; 12: 187–193.
- Bertram M, Waidhas F, Jevric M, Fromm L, Schuschke C, Kastenmeier M, Görling A, Moth-Poulsen K, Brummel O, Libuda J. Norbornadiene photoswitches anchored to well-defined oxide surfaces: From ultrahigh vacuum into the liquid and the electrochemical environment. J Chem Phys. 2020; 152: 044708.
- Ree N, Mikkelsen KV. Benchmark study on the optical and thermochemical properties of the norbornadiene-quadricyclane photoswitch. Chemical Physics Letters. 2021; 779: 138665.
- Zen-ichi Y. Journal of Photochemistry. 1985; 29: 27-40.
- Yanai T, Tew DP, Handy NC. A new hybrid exchange–correlation functional using the Coulomb-attenuating method (CAM-B3LYP). Chemical Physics Letters. 2004; 393: 51–57.
- Tawada Y, Tsuneda T, Yanagisawa S, Yanai T, Hirao K. A long-range-corrected time-dependent density functional theory. J Chem Phys. 2004 May 8;120(18):8425-33. doi: 10.1063/1.1688752. PMID: 15267767.
- Zhao Y, Truhlar DG. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theoretical Chemistry Accounts. 2008; 120: 215–241.
- Yu HS, He X, Li SL, Truhlar DG. MN15: A Kohn-Sham global-hybrid exchange-correlation density functional with broad accuracy for multi-reference and single-reference systems and noncovalent interactions. Chem Sci. 2016 Aug 1;7(8):5032-5051. doi: 10.1039/c6sc00705h. Epub 2016 Apr 6. Erratum in: Chem Sci. 2016 Sep 1;7(9):6278-6279. PMID: 30155154; PMCID: PMC6018516.
- Peverati R, Truhlar DG. Communication: A global hybrid generalized gradient approximation to the exchange-correlation functional that satisfies the second-order density-gradient constraint and has broad applicability in chemistry. J Chem Phys. 2011 Nov 21;135(19):191102. doi: 10.1063/1.3663871. PMID: 22112059; PMCID: PMC3248024.
- Chai JD, Head-Gordon M. Long-range corrected hybrid density functionals with damped atom-atom dispersion corrections. Phys Chem Chem Phys. 2008 Nov 28;10(44):6615-20. doi: 10.1039/b810189b. Epub 2008 Sep 29. PMID: 18989472.
- Curtiss LA, McGrath MP, Blaudeau J, Davis NE, Binning RC, Radom L. Extension of Gaussian‐2 theory to molecules containing third‐row atoms Ga–Kr. The Journal of Chemical Physics. 1995; 103: 6104–6113.
- Blaudeau JP, McGrath MP, Curtiss LA, Radom L. Extension of Gaussian-2 (G2) theory to molecules containing third-row atoms K and Ca. The Journal of Chemical Physics. 1997; 107: 5016–5021.
- Wachters AJH. Gaussian Basis Set for Molecular Wavefunctions Containing Third‐Row Atoms. The Journal of Chemical Physics. 1970; 52: 1033–1036.
- Hay PJ. Gaussian basis sets for molecular calculations. The representation of 3d orbitals in transition‐metal atoms. The Journal of Chemical Physics. 1977; 66: 4377–4384.
- McLean AD, Chandler GS. Contracted Gaussian basis sets for molecular calculations. I. Second row atoms, Z=11–18. The Journal of Chemical Physics. 1980; 72: 5639–5648.
- Krishnan R, Binkley JS, Seeger R, Pople JA. Self‐consistent molecular orbital methods. XX. A basis set for correlated wave functions. The Journal of Chemical Physics. 1980; 72: 650–654.
- Raghavachari K, Trucks GW. Highly correlated systems. Excitation energies of first row transition metals Sc–Cu. The Journal of Chemical Physics. 1989; 91: 1062–1065.
- McGrath MP, Radom L. Extension of Gaussian‐1 (G1) theory to bromine‐containing molecules. The Journal of Chemical Physics. 1991; 94: 511–516.
- Weigend F, Ahlrichs R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Physical Chemistry Chemical Physics. 2005; 7: 3297–3305.
- Weigend F. Accurate Coulomb-fitting basis sets for H to Rn. Physical Chemistry Chemical Physics. 2006; 8: 1057–1065.
- Frisch MJ. Gaussian˜16 Revision C.01. 2016; Gaussian Inc. Wallingford CT.
- Cancès E, Mennucci B, Tomasi J. A new integral equation formalism for the polarizable continuum model: Theoretical background and applications to isotropic and anisotropic dielectrics. The Journal of Chemical Physics. 1997; 107: 3032–3041.
- Jacovella U, Carrascosa E, Buntine JT, Ree N, Mikkelsen KV, Jevric M, Moth-Poulsen K, Bieske EJ. Photo- and Collision-Induced Isomerization of a Charge-Tagged Norbornadiene-Quadricyclane System. J Phys Chem Lett. 2020 Aug 6;11(15):6045-6050. doi: 10.1021/acs.jpclett.0c01198. Epub 2020 Jul 15. PMID: 32539402; PMCID: PMC7416310.
- Kendall RA, Dunning TH, Harrison RJ. Electron affinities of the first‐row atoms revisited. Systematic basis sets and wave functions. The Journal of Chemical Physics. 1992; 96: 6796–6806.
- Woon DE. Dunning TH. aussian basis sets for use in correlated molecular calculations. III. The atoms aluminum through argon. The Journal of Chemical Physics. 1993; 98: 1358–1371.
- Jmol: an open-source Java viewer for chemical structures in 3D. http://www.jmol.org/.
- Mansø M , Petersen AU , Moth-Poulsen K , Nielsen MB . Establishing linear-free-energy relationships for the quadricyclane-to-norbornadiene reaction. Org Biomol Chem. 2020 Mar 18;18(11):2113-2119. doi: 10.1039/d0ob00147c. PMID: 32119025.